Die Rolle der EU bei der Zulassung und Beschaffung von Covid-19-Impfstoffen
In den nächsten Wochen und Monaten soll es in der EU mehrere zugelassene Impfstoffe gegen Covid-19 geben. Die Europäische Arzneimittel-Agentur (EMA) arbeitet eng mit Entwicklerinnen und Entwickler potenzieller Covid-19-Impfstoffe sowie Regulierungspartnerinnen und -partner zusammen und mobilisiert ihre eigenen Ressourcen, um sicherzustellen, dass sichere und wirksame Impfstoffe so schnell wie möglich die Bevölkerung erreichen.
Die EMA spielt eine wichtige Rolle bei der Entwicklung, wissenschaftlichen Bewertung, Zulassung und Überwachung von Covid-19-Impfstoffen in der Europäischen Union. Zusammen mit dem europäischen Arzneimittelregulierungsnetzwerk unterstützt die EMA die Entwicklung von Impfstoffen und hat Schritte unternommen, um die Bewertungsprozesse für diese Impfstoffe zu beschleunigen. Angesichts der Pandemie leiten die EMA und die Aufsichtsbehörden in Europa Ressourcen um, um Prozesse zu beschleunigen und die Fristen für die Bewertung und Zulassung von Covid-19-Impfstoffen zu verkürzen.
Videokonferenz zwischen Kommissionspräsidentin von der Leyen und Bundeskanzler Kurz: Koordinierte Impfstoff-Beschaffung ist großer Erfolg der EU
Durch die Impfung gebe es "Licht am Ende des Tunnels und eine Rückkehr zur Normalität im Jahr 2021", betonte Bundeskanzler Sebastian Kurz bei einer Videokonferenz mit Kommissionspräsidentin Ursula von der Leyen am 27. November 2020. Der Beschaffungsprozess der Covid-19-Impfstoffe sei "gut aufgestellt" und könne als eine der großen Erfolgsgeschichten der EU im Gesundheitsbereich betrachtet werden. Für die Verhandlungen und den Abschluss der Vorkaufverträge mit den potenziellen Herstellern standen der EU insgesamt 2,7 Milliarden Euro zur Verfügung. 2 Milliarden Impfdosen konnte die Europäische Kommission sicherstellen; davon stünden 2 Prozent Österreich zu. Die Impfung selbst müsse zweimal durchgeführt werden und sei in Österreich für die Bürgerinnen und Bürger, die sich impfen lassen möchten, kostenlos. Die EU-Kommission habe bei der Beschaffung der Impfstoffe Überkapazitäten eingeplant, um die Nachbarschaft der EU zu unterstützen, insbesondere die Westbalkan-Staaten als potenzielle Empfänger. Bundeskanzler Kurz versicherte, dass in der EU und in Österreich nur ein hundertprozentig sicherer Impfstoff zugelassen werde.

Bereits 2 Anträge auf Zulassung gestellt
In den nächsten Wochen und Monaten (Stand 21. Dezember 2020) sollen die ersten in der EU zugelassenen Impfstoffe verfügbar sein. Die Europäische Arzneimittelagentur (EMA) hat am 1. Dezember 2020 sowohl von BioNTech/Pfizer als auch Moderna Anträge auf die bedingte Marktzulassung der Impfstoffe erhalten, welche die Unternehmen gegen Covid-19 entwickelt haben. Die EMA hat ihre Prüfung des COVID19-Impfstoffs von BioNTech/Pfizer abgeschlossen und am 21. Dezember 2020 seine Zulassung in der EU empfohlen. Nach einer formellen Prüfung der Marktzulassung durch die Europäische Kommission starten die Impfungen EU-weit im Zeitraum von 27. bis 29. Dezember 2020.
In der EMA wurde eine multidisziplinäre "Covid-19 Task Force" (ETF) eingerichtet, in welcher Schlüsselexpertinnen und -experten aus dem gesamten europäischen Netzwerk für Arzneimittelregulierungen zusammenkommen, um eine schnelle und koordinierte Reaktion auf die Pandemie sicherzustellen. Auch die EU-Mitgliedstaaten treffen umfassende Vorbereitungen für die Planung, Logistik und Durchführung der Impfungen. Das Ziel der EU-Kommission ist, innerhalb von 12 bis 18 Monaten einen Impfstoff zu finden, obwohl das normalerweise 10 Jahre dauert.
Keine Abstriche bei der Qualitätssicherung
Die EMA und ihre Partnerinnen und Partner stellen sicher, dass für alle Impfstoffe dieselben hohen regulatorischen Standards hinsichtlich Qualität, Sicherheit und Wirksamkeit gelten und auch erfüllt werden. Covid-19-Impfstoffe können nur zugelassen und verwendet werden, wenn sie alle Anforderungen erfüllen, die in der EU-Arzneimittelgesetzgebung festgelegt sind. Die EU-Arzneimittelgesetzgebung stellt sicher, dass Impfstoffe erst zugelassen werden, nachdem die wissenschaftliche Bewertung gezeigt hat, dass ihr Gesamtnutzen die Risiken überwiegt. Die Vorteile eines Impfstoffs beim Schutz von Menschen vor Covid-19 müssen demnach weitaus größer sein als alle Nebenwirkungen oder potenziellen Risiken.
EMA-Zulassungsverfahren für Impfstoffe im Detail
Als ersten Schritt, um die Zulassung eines Impfstoffs in der EU zu erhalten, legt der Impfstoffentwickler die Ergebnisse aller Tests beziehungsweise Untersuchungen den Arzneimittelaufsichtsbehörden in Europa vor. Dies ist Teil eines Zulassungsantrags.
Der "Ausschuss für Humanarzneimittel" (CHMP) der EMA bewertet anschließend alle Anträge für Covid-19-Arzneimittel innerhalb des Mindestzeitraums, der erforderlich ist, um die Vorteile und Risiken des Arzneimittels zu beurteilen. Dabei bewertet die EMA die Anträge gemäß ihren üblichen Standards, allerdings anhand eines fortlaufenden Überprüfungsverfahrens ("Rolling Review"). Die "Rolling Review" dient dazu, das Verfahren zur Zulassung eines Covid-19-Impfstoffes zu beschleunigen. Dafür wird mit der Bewertung von Datenpaketen der nichtklinischen und klinischen Entwicklung eines Impfstoffkandidaten bereits begonnen, bevor alle erforderlichen Daten für einen Zulassungsantrag erhoben sind. Die EMA stellt dabei sicher, dass die wissenschaftlichen Expertinnen und Experten, welche die Arzneimittel bewerten, keine finanziellen oder sonstigen Interessen haben, die ihre Unparteilichkeit beeinträchtigen. Das Komitee setzt sich aus Vertreterinnen und Vertretern der einzelnen Mitgliedstaaten zusammen, die diese Unterlagen begutachten und mit den Firmen auch über die Datenlage diskutieren. Um die Unabhängigkeit der Evaluierungen zu gewährleisten, ist die wissenschaftliche Evaluierungsarbeit der EMA der Öffentlichkeit zugänglich.
Die fortlaufende Überprüfung der Impfstoffe beginnt mit der Auswertung der ersten Datenmengen durch CHMP und wird fortgesetzt, bis genügend Beweise für einen formellen Zulassungsantrag für das Inverkehrbringen vorliegen. Die EMA kann erst dann zu einer Schlussfolgerung über die Sicherheit und Wirksamkeit dieser Impfstoffe gelangen, wenn der Entwickler diese Nachweise vorgelegt hat und diese von EMA bewertet wurden.
Zu diesen Nachweisen gehören Ergebnisse aus bereits laufenden groß angelegten klinischen Studien, die Aufschluss darüber geben, wie wirksam die Impfstoffe beim Schutz der Menschen vor Covid-19 sind. Bei diesen großen randomisierten kontrollierten klinischen Studien werden Freiwillige nach dem Zufallsprinzip ausgewählt, um den getesteten Impfstoff zu erhalten, und unter kontrollierten Bedingungen gemäß strengen Protokollen überwacht. Die randomisierte kontrollierte Studie (randomized controlled trial, RCT) ist die hochwertigste Form einer klinischen Studie.
Klinische Studien durchlaufen 3 einzelne Studie beziehungsweise 3 Phasen:
- Während Phase 1 testen Forscherinnen und Forscher im Allgemeinen ein neues mögliches Medikament bei gesunden Menschen. In den meisten Fällen nehmen 20 bis 80 Freiwillige an Phase 1 teil. Der Hauptzweck von Phase-1-Studien ist, die Sicherheit eines neuen Medikaments zu bewerten, bevor weitere Studien durchgeführt werden. Zusätzlich zur Sicherheit können Forscherinnen und Forscher in einer Phase-1-Studie weitere Fragen beantworten, die sich darauf beziehen, wie viel Arzneimittel nach der Verabreichung im Blut gemessen wird, wie das Arzneimittel im Körper wirkt und welche Nebenwirkungen mit einer erhöhten Dosierung verbunden sind.
- In Phase 2 verabreichen Forscherinnen und Forscher das Arzneimittel einer größeren Gruppe von Patientinnen und Patienten mit der Krankheit (typischerweise bis zu 100), um zunächst seine Wirksamkeit zu bewerten und seine Sicherheit weiter zu untersuchen. Ein Schwerpunkt der Phase 2 liegt auf der Bestimmung der optimalen Dosis oder Dosen eines Medikaments, um zu bestimmen, wie das Medikament am besten verabreicht werden kann, um den möglichen Nutzen zu maximieren und gleichzeitig die Risiken zu minimieren.
- Das Hauptaugenmerk der Phase 3 liegt auf dem Nachweis der Wirksamkeit und Sicherheit des Impfstoffs. In dieser Phase sind in der Regel 300 bis 3.000 Patientinnen und Patienten beteiligt, für die das Arzneimittel möglicherweise verwendet werden soll. Die Teilnehmerinnen und Teilnehmer erhalten entweder das zu bewertende Medikament oder sind Teil einer Kontrollgruppe und erhalten entweder den aktuellen Behandlungsstandard oder ein Placebo.
Wenn eine Behandlung in einer Phase erfolgreich ist, geht sie zur nächsten Phase über.
Die Bewertung umfasst alle verfügbaren Daten zur Sicherheit der Impfstoffe, die sich aus diesen Studien ergeben und Daten zu deren Qualität, etwa Informationen zu den Inhaltsstoffen der Impfstoffe und zu deren Herstellung. Die EMA kann den Gesamtzeitplan für die Bewertung der Covid-19-Impfstoffe nicht vorhersagen, erwartet jedoch, dass der Prozess kürzer ist als der Prozess einer Standard-Bewertung. Dies ist dank der Zeit möglich, die während der fortlaufenden Überprüfung eingespart wurde.
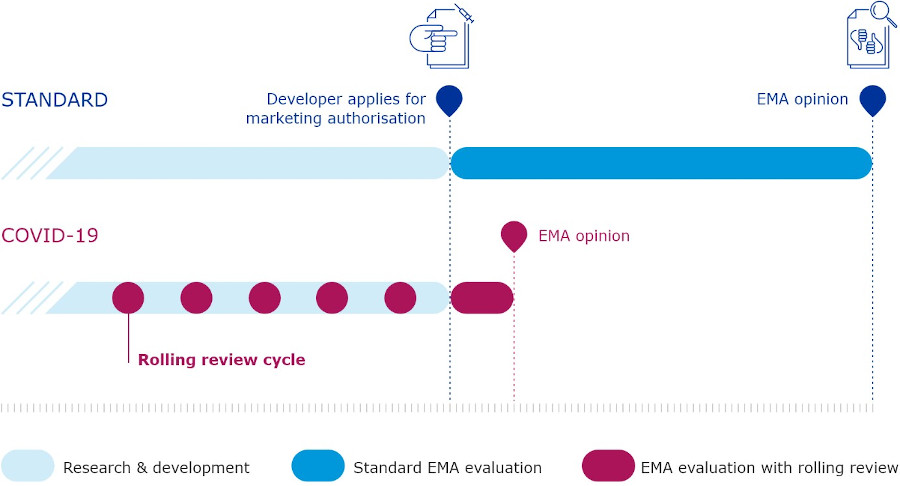
Sobald die EMA einen positiven Bescheid erlässt ist die Europäische Kommission für die Zulassung für alle EU-Mitgliedsstaaten zuständig.
In der Zeit nach der Autorisierung stellt die EMA sicher, dass die genehmigten Impfstoffe weiterhin genutzt werden können. Das heißt, es wird weiter untersucht, ob es bei den Testpersonen während der kommenden Zeit zu Impfstoffnebenwirkungen kommt. Auch die Langzeitwirkung wird immer wieder überprüft, etwa indem gemessen wird, ob geimpfte Personen immer noch Abwehrstoffe im Blut haben. Diese Überwachung der Testpersonen wird durch die Meldungen vermuteter Nebenwirkungen bei geimpften Patienten aus der Bevölkerung und Personen aus den Gesundheitsberufen ergänzt. Zu diesem Zweck gibt es EU-weit ein umfassendes System für die Sicherheitsüberwachung (Pharmakovigilanz-System). Sämtliche Meldungen vermuteter Nebenwirkungen werden EU-weit in einer Datenbank namens "EudraVigilance" (European Union Drug Regulating Authorities Pharmacovigilance) gesammelt und ausgewertet. Darüber hinaus werden neue Studien gestartet. Sie sollen mögliche seltene Nebenwirkungen in der Bevölkerung aufdecken und helfen, die Wirksamkeit in unterschiedlichen Gruppen, etwa bei Älteren oder Menschen mit Vorerkrankungen, im Auge zu behalten.
Ausnahme einer bedingten Zulassung
Im Falle von Covid-19-Impfstoffen soll es bei bisher gestellten Anträgen die Empfehlung einer bedingten Zulassung geben, welche dringliche medizinische Bedürfnisse befriedigen soll. Wenn der Nutzen das Risiko überwiegt, das von weniger als normalerweise erforderlichen Daten ausgeht, kann den Antragstellern im Interesse der öffentlichen Gesundheit eine bedingte Genehmigung gegeben werden. Fehlende Daten müssen allerdings so schnell wie möglich nachgereicht werden. Diese bedingte Zulassung gilt für ein Jahr und kann anschließend verlängert werden.
Aufstellung von Risikomanagementplänen
Die EMA hat in Fall von Covid-19-Impfstoffen Risikomanagementpläne aufgestellt. Diese legen fest, wie Entwickler-Unternehmen die Sicherheit ihrer Impfstoffe überwachen, darüber Bericht erstatten und wie Risiken nach der Zulassung eines Covid-19-Impftsoffes charakterisiert und verwaltet werden.
Unternehmen müssen zusätzlich zu regelmäßigen Sicherheitsaktualisierungsberichten monatliche Zusammenfassungen ihrer Sicherheitsberichte einreichen und Prozesse einrichten, um ein hohes Volumen an Sicherheitsberichten zu verwalten. Sie sind auch dazu verpflichtet, weitere Studien zu ihren bereits genehmigten Impfstoffen durchführen.
Weitere Informationen
- Das Europäische Arzneimittelregulierungssystem, EMA
- Covid-19-Impfungen: Entwicklung, Evaluierung, Autorisierung und Überwachung, EMA, November 2020 (Englisch)
- Pressemitteilung zur Task Force, EMA, 9. April 2020 (Englisch)
- Kernanforderungen der Risikomanagementpläne, EMA, November 2020 (Englisch)
- Pressemitteilung über den Antrag auf bedingte Genehmigung durch BioNTech und Pfizer, EMA, 1. Dezember 2020 (Englisch)
- Pressemitteilung über den Antrag auf bedingte Genehmigung durch Moderna, EMA, 1. Dezember 2020 (Englisch)
- Pressemitteilung zur Beantragung der Zulassung von BioNTech/Pfizer und Moderna Covid-19-Impfstoffen, EU-Kommission Vertretung in Deutschland, 1. Dezember 2020
- Pressemitteilung über die Prüfung des BioNTech Impfstoff bis 21. Dezember 2020, EU-Kommission Vertretung in Deutschland, 15. Dezember 2020
- Corona-Informationen auf der Seite des Sozialministeriums
- Coronavirus response, Website der Europäischen Kommission